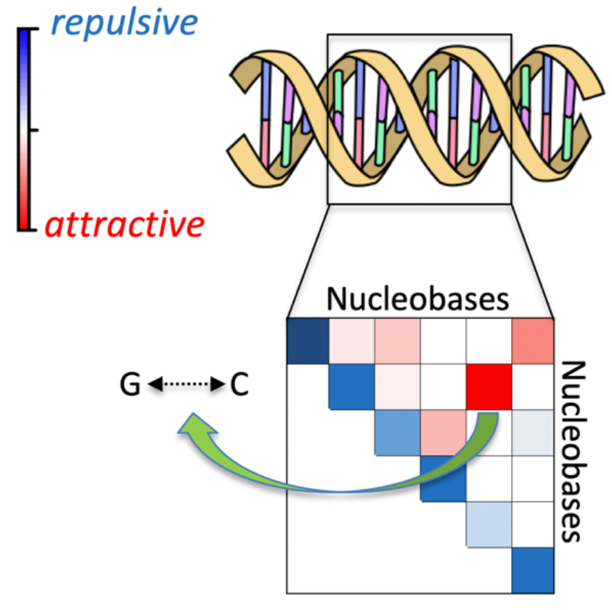
Charge densities and London dispersion forces

Prof. Katharine L. C. Hunt
Department of Chemistry, Michigan State University, East Lansing, Michingan, USA
Abstract
In 1939, Feynman published without proof a statement that the dispersion force between two atoms in S states comes from the attraction of each nucleus to the distorted electronic charge distribution in its own vicinity. In 1990, Hunt proved Feynman’s statement by use of perturbation theory within the polarization approximation and extended the result to molecules of arbitrary symmetry. Later work has raised question about this interpretation. In 2019, Kooi and Gori-Giorgi developed a density-functional theory that yields the dispersion energy with high accuracy, with no change in the electronic charge density at any order. Calculations on He and Ar atom pairs show that the electron density drops in the immediate vicinity of each nucleus, within the van der Waals bonding region. My group has derived dispersion forces with high accuracy from the correlation contribution to the charge density of the hydrogen molecule in the lowest triplet state at long range. As found previously, the electron density drops in the immediate vicinity of each nucleus, within the bonding region. However, the electron density that produces the forces on the nuclei rises in the same region, validating Feynman’s statement.
Capturing Noncovalent Interactions in Molecular String Representations

Prof. Robert Pollice
Stratingh Institute for Chemistry, University of Groningen, Groningen, The Netherlands
Abstract
In this talk, I will introduce about our current work on developing a molecular string representation that not only captures covalent bonds and other strong interactions, but also noncovalent interactions. I will discuss the theoretical foundation and show application examples both from a cheminformatics perspective and with respect to machine learning models. As outlook, I will discuss future extensions towards a general framework allowing to create custom representations from any structure on demand.
Engineering Functional Organic Materials through Noncovalent Interactions

Prof. Ganna Gryn’ova
School of Chemistry, University of Birmingham, Birmingham, United Kingdom
Abstract
Gregor Lauter1, Catherine Mollart1, Michelle Ernst2, Ganna Gryn’ova12
1School of Chemistry, University of Birmingham, B15 2TT Birmingham, United Kingdom
2Institute for Geological Sciences, University of Bern, 3012 Bern, Switzerland
Functional, topologically complex organic systems are rising stars in modern materials science due to their biocompatibility, structural variability, and wealth of physico-chemical properties. Their practical applications often involve interactions with small molecular targets (e.g., gases, environmental pollutants, and drugs) via relatively weak non-covalent forces. In this contribution we will discuss our recent efforts towards reliable yet scalable computational approaches to modelling, quantifying, and analysing the interactions between functional organic materials and their small molecule targets. We focus on two types of systems: (1) graphene-based materials for sensing and catalysis, and (2) organic frameworks for molecular storage. We will highlight the methodological tips and tricks for modelling the relevant host-guest complexes using multiscale approaches at an optimal ratio of accuracy and computational cost and illustrate how intermolecular forces can guide targeted design of novel catalysts and cages.
Understanding London Dispersion Effects in Molecular Chemistry with Local Coupled Cluster Methods

Prof. Giovanni Bistoni
Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Perugia, Italy
Abstract
Local correlation techniques exploit the short-range nature of dynamic electron correlation to mitigate the inherent steep scaling of wavefunction-based methods. In this contribution, I discuss novel methods[1-5] based on local coupled cluster theory for studying dispersion effects on reactivity[6] and interactions[7] of large and complex chemical systems (Fig. 1). The family of local correlation methods offers a powerful toolbox for achieving both high accuracy and chemical insights simultaneously.
1. G. Bistoni, WIREs Comput Mol Sci. 2020; 10:e1442
2. G. Bistoni, A. Altun, Z. Wang, F. Neese. Acc. Chem. Res. 2024, 57, 9, 1411
3. A. Altun, I. F. Leach, F. Neese, G. Bistoni, Angew. Chem. Int. Ed. 2025, 64, e202421922.
4. Gianluca Regni, Lorenzo Baldinelli, G. Bistoni, Submitted
5. Martina Colucci, Gianluca Regni, Christoph Riplinger, Frank Neese, G. Bistoni, in preparation
6. R. Kayal, L. Baldinelli, I. Harden, F. Neese, G. Bistoni, Chem. Sci. 2025, 16, 2700.
7. A. Altun, M. Garcia-ratés, F. neese, G. Bistoni, Chem. Sci., 2021, 12, 12785-12793
Non-covalent interactions in Real Space: Tracing Electron Correlation, Intermolecular Bonds, and Dispersion from the IQA Perspective

Prof. Ángel Martín Pendás
Facultad de Química, Universidad de Oviedo, Oviedo, Spain.
Abstract
A fundamental understanding of dispersion and other non-covalent interactions demands a framework that captures correlation energies with spatial resolution. Actually, many of the most successful computational frameworks devised to include dispersion corrections in a computationally efficient way, such as the DFT-D3(4), MBD, or XDM formalisms (see Ref. [1] for a survey), use some atomic partitioning. Here we show how to apply the Interacting Quantum Atoms (IQA) formalism [2,3], derived from real-space QTAIM [4] partitions, to investigate the nature of non-covalent interactions as well as the localization of electron correlation energy across a variety of molecular systems and intermolecular complexes. Leveraging high-level correlated wavefunctions, including CCSD(T) and near-exact FCI densities, we demonstrate that intra-atomic contributions overwhelmingly dominate the correlation energy, while interatomic terms provide chemically meaningful insight into both short-range and long-range correlation [5,6]. We show that the sign and magnitude of interatomic correlation energies offer a robust criterion to distinguish covalent from non-bonded or dispersive interactions, allowing a real-space reinterpretation of dispersion forces. A focused analysis of the dihydrogen triplet state across varying internuclear separations reveals that dispersion-like behavior emerges naturally in the interatomic correlation terms at large distances, while short-range contributions dominate near equilibrium. Our results propose the interatomic correlation energy descriptor as a reliable, position-space marker of dispersion, opening the door to intramolecular dispersion definitions within real-space frameworks. The combination of IQA energy decomposition with relaxed density matrices presents a powerful tool for dissecting non-covalent interactions in complex chemical environments and may inform the development of dispersion-aware real-space force fields.
[1] Non-Covalent Interactions in Quantum Chemistry and Physics. Alberto Otero de la Roza, Gino A. DiLabio, Eds. Elsevier (2017). ISBN 9780128098356
[2] M. A. Blanco, A. Martín Pendás, E. Francisco, J. Chem. Theory Comput. 1, 1096–1109 (2005)
[3] A. Martín Pendás, E. Francisco, Nat Commun 13, 3327 (2022).
[4] R. F. W. Bader. Atoms in molecules, Oxford University Press (1991). ISBN 978-0198551683
[5] J. L Casals-Sainz, J. M. Guevara-Vela, E. Francisco, T. Rocha-Rinza, and A. Martín Pendás, ChemPhysChem 18, 3553-3561 (2017)
[6] F. Jiménez-Grávalos, M. Gallegos, A. Martín Pendás, A. S. Novikov, J. Comput. Chem. 42, 676-687 (2021)
A Gravitational-Like Behavior of Dispersion Interactions1

Prof. Sason Shaik
Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel.
Abstract
We present computational results of many-body dispersion (MBD) interactions for more than 40 pairs of molecular and atomic species: hydrocarbons, silanes, corresponding fluorinated derivatives, pairs of CnHn cages, pairs which have multiple H---H contacts between the molecules, as well as pairs having π−π interactions, and pairs of noble gases. The calculations reveal that the MBD stabilization energy (EDISP,MBD) obeys a global relationship, which is gravitational-like. It is proportional to the product of the masses of the two molecules (M1*M2) and inversely proportional to the corresponding distances between the molecular centers-of-mass (RCOM‑COM). This relationship reflects the interactions of instantaneous dipoles, which are generated by the ensemble of bonds/atoms in the interacting molecules. Using Grimme’s D4 corrected dispersion energy (EDISP,D4), we find that the data sets for EDISP,MBD and EDISP,D4 are strongly correlated (r2= 0.997). Based on valence-bond modeling, the intra- and inter-molecular dispersion interactions occur primarily due to the increased contributions of the oscillating-ionic VB structures which maintain favorable electrostatic interactions; the. [RC+:H−+H :C−R] and [RC:−+H −H:C+R] structures; R symbolizes a general residue. The local charges (on the C and H atoms) are propagated to the entire ensemble of bonds/atoms in the R residues, thus bringing about the gravitational dependence of dispersion.
1. D. Danovich, A. Tkachenko, S. Alvarez, S. Shaik, J. Am. Chem. Soc. 146, 311204 (2024)
Leveraging the Many-Body Dispersion Model: Accessing Charge Density Distortions and Quantum Electrodynamics Properties in Large Molecular Systems

Dr. Matteo Gori
Department of Physics and Materials Science, University of Luxembourg, Luxembourg City, Luxembourg.
Abstract
The Many-Body Dispersion (MBD) model is a highly accurate and effective approach for describing many-body van der Waals interactions in post-DFT calculations. Unlike other methods with similar goals, the MBD model characterizes the electronic response properties of the charge density field using a set of quantum Drude oscillators—quantum harmonic oscillators that carry an electric dipole. This representation has the potential to capture additional aspects of the electronic structure beyond many-body dispersion energy corrections, particularly in large molecular systems such as realistic biomolecular complexes.
Motivated by this picture, we present a twofold investigation into the capabilities of the MBD model for describing the geometry of electronic quantum charge distributions and localized (paramagnetic) currents in large molecular systems. First, we demonstrate how the MBD model can be employed to capture distortion effects on the ground-state charge density induced by many-body dispersion interactions, with potential applications in density-corrected DFT and in the visualization of dispersion interactions within large molecules. Second, we explore how the MBD model can be embedded within the established molecular Quantum Electrodynamics (QED) framework, offering an effective representation of the polarization and magnetization field operators in large molecular systems. This integration opens new avenues for applications such as polariton chemistry in QED cavities and chiral discrimination interactions in biomolecular systems.
The Functional Group Correction approach versus machine learning

Prof. Berta Fernández Rodríguez
Departamento de Química Física, Facultade de Química, Universidade de Santiago de Compostela, Santiago de Compostela, Spain.
Abstract
In recent work we developed the functional group correction (FGC) approach to correct the description of intermolecular interactions given by semiempirical quantum mechanical methods; in particular, we applied it to the PM6 and the GFN2-xtb approaches [1]. The new method is based on pairwise analytical corrections that work for specific functional groups and are derived from fits to interaction energy differences between the B3LYP-D3/def2-TZVP DFT values, taken as reference, and those provided by the particular semiempirical method [2]. Previously obtained corrections performed outstandingly well for alkanes and alkenes. In general, for the systems included in training and validation sets, the errors obtained with the PM6-FGC and xTB-FGC methods are within chemical accuracy. Considering the simplicity of the FGC approach and the speed of the involved calculations, its performance and outcome are promising, with results comparable to those provided by a recently developed state-of-the art delta machine learning (delta-ML) methodology, i.e. the PM6-ML approach [3], and significantly better than those obtained using the AIQM2 method [4].
[1] E. M. Cabaleiro-Lago, B. Fernández, R. Rodríguez-Fernández, J. Rodríguez-Otero, S. A. Vázquez, J. Chem. Phys., 2023, 158, 124105.
[2] Pérez-Tabero, B. Fernández, E. M. Cabaleiro-Lago, E. Martínez-Núñez, S. A. Vázquez, J. Chem. Theory Comput. 2021, 17, 5556-5567.
[3] M. Nováček, J. Řezác, Journal of Chemical Theory and Computation, 2025, 21 (2), 678-690.
[4] Y. Chen, P. O. Dral, 2024. Preprint on ChemRxiv: https://chemrxiv.org/engage/chemrxiv/article-details/67042664cec5d6c1427a144f (2024-10-08).
Unraveling the Nuclear Motion of Weakly Bound Materials

Prof. Mariana Rossi
MPI for the Structure and Dynamics of Matter, Hamburg, Germany.
Abstract
Weakly bonded interfaces composed by molecular and solid-state inorganic materials give rise to a rich variety of nuclear motion and tunable nuclear structure that is tightly connected to diverse electronic properties in these systems. In my talk, I will discuss the importance of accounting for nuclear motion to study diverse properties of these systems, and the importance of doing so. I will show how we push the limits of density-functional theory and different ab initio techniques that capture nuclear motion to unravel the properties of realistic interfaces [1].
I will discuss how they can be connected to first-principles electronic structure and machine-learning approaches [2,3]. Applications where the quantum nature of the nuclei become indispensable to assess structural and electronic properties these interfaces will be shown and discussed [4], as well as how these can be characterised by the simulation of experimentally observable quantities like tunneling rate constants and advanced vibrational spectroscopy [5,6].
[1] J. Chem. Phys. 154, 170902 (2021);
[2] J. Chem. Theory Comput. 17, 7203- 7214 (2021);
[3] J. Chem. Phys. 159, 014103 (2023);
[4] arXiv:2411.10994 (2024)
[5] J. Chem. Phys. 156, 194106 (2022);
[6] J. Phys. Chem. Lett. 14, 6850 (2023)
Predicting Reactivity from Density

Prof. Jean Pierre Djukic
Laboratoire de Chimie et Systémique Organométallique, Strasbourg, France.
Abstract
Sara Figueireido de Alcantara Morais (a), Yann Cornaton,(a) Eric Hénon(b) and Jean-Pierre Djukic (a),*
(a) Laboratoire de Chimie et Systémique Organométallique, UMR 7177, 4 rue Blaise Pascal 67000 Strasbourg, France.
(b) Institut de Chimie Moléculaire de Reims, UMR CNRS 7312, Université de Reims - BP 1039, F-51687 Reims Cedex 2, France
Local non-covalent interactions may be informative a potential propensity of a molecule to undergo intramolecular reactions. In the Independent Gradient Model (IGM)[1] introduced by Hénon and coworkers, local non-covalent interactions are the weakest type of interatomic interactions. Yet, their assessment by this analytical method in crucial reactant complexes gives unique insights into the changes of electronic structure occurring during reactions occurring within the coordination sphere of a transition metal complex.[2,3] Extending the use of IGM to the analysis of full intrinsic reaction energy profiles gives access to detailed sequences of reaction events that otherwise could not be sensed.[4] These reaction events are all characterized by remarkable transient structures that correspond to key transformations involving electron density contributions from surrounding atomic centers. This new approach of reactivity dynamics is particularly useful for the analysis of concerted reactions relevant to catalysis.[4] It allows the monitoring of the electronic influence of non-central atomic centers in key events that was long inaccessible making the engineering of reaction more dependent on intuition.
1) J. Comput. Chem. 2023, 44(20), 1750. https://doi.org/10.1002/jcc.27123
2) Acc. Chem. Res. 2021, 54, 3828–3840
3) Synlett 2023, 34, 1169-1173
4) Manuscript(s) in preparation.
Van der Waals effects in surface and interface chemistry

Prof. Wei Liu
Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin, China.
Abstract
In the fields of catalysis and new materials research, surface and interfacial interactions play a crucial role in influencing various fundamental physicochemical processes. Among these, accurately describing many-body van der Waals (vdW) interactions remains a key challenge in achieving precise control over such processes. In this talk, I will present recent progress from our group in addressing this challenge: we developed a method to accurately describe many-body vdW forces, uncovering the fundamental nature of many-body effects, including screening, polarization, and their coupling with chemical bonding. Based on this approach, we elucidated the decisive role of screening effects in the formation of Langmuir precursor states, identified 56 new systems where physical and chemical adsorption coexist, and enabled the precise recognition of chiral molecules on defective surfaces, as well as the accurate prediction of reaction activity in prototypical electrocatalytic materials. This work opens new avenues for the rational design of advanced materials with tailored surface and interfacial properties, paving the way for further breakthroughs in energy conversion, catalysis, and materials engineering.
[1] G. Cao, S. Yang, J.-C. Ren*, and W. Liu*, Electronic Descriptors for Designing High-Entropy Alloy electrocatalysts by Leveraging Local Chemical Environments, Nat. Commun. 16 (2025) 1251.
[2] C. Chen, Y. Ma, K. Yao, Q. M. Ji, W. Liu*, Enantioselective adsorption on chiral ceramics with medium entropy, Nat. Commun. 15 (2024) 10105.
[3] J. Zhou, S. Yang, Y. Zhang, J.-C. Ren*, and W. Liu*, An effective descriptor for screening single-molecule conductance switches, J. Am. Chem. Soc. 146 (2024) 6962-6973.
[4] M. Fang, J. Han, S. He, J.-C. Ren*, S. Li*, and W. Liu*, Effective screening descriptor for MXenes to enhance sulfur reduction in lithium-sulfur batteries, J. Am. Chem. Soc. 145 (2023) 12601-12608.
Enabling gold standard level of accuracy in modeling complex noncovalent interactions between large (bio)molecules

Prof. Péter R. Nagy
Department of Physical Chemistry and Materials Science, Faculty of Chemical Technology and Biotechnology, Budapest University Of Technology And Economics, Budapest, Hungary
Abstract
The accurate modeling of various ionic, aromatic, polarized, sigma-hole, surface, etc. interactions and especially their coupling to each other and to reactive processes remains challenging. We report advances in accelerating coupled-cluster (CC) methods and their convergence to the complete basis set limit (CBS), providing chemical accuracy even for large systems of complicated (single-reference) electronic structure.
Our CC methods with state-of-the-art accuracy-over-cost performance include frozen and local natural orbital (FNO & LNO) approaches, optimized parallel closed- and open-shell implementations, linear-scaling MP2, LNO-CCSD(T), and general-order LNO-CC, as well as (so far conventional) CC gradients [reviewed in 1,2]. To improve basis set convergence, we developed an MPI+OpenMP parallel FNO-CCSD(F12*)(T+) code, a generally applicable floating orbital Gaussian basis (FOG) approach[2,3], and an LNO-accelerated DFT-based basis set correction[4].
LNO-CCSD(T)/CBS energies usually take about 1-2 order of magnitude more time than HF/hibrid DFT in the same basis set, have exceptionally low minimal memory and disk requirement (10s-100 GB), and are accompanied with robust uncertainty estimates. This makes them routinely applicable for molecules of 100s of atoms even with a single node.[1,2]
We utilize the efficiency of LNO-CCSD(T) to model supramolecular and protein-drug/ion complexes, interactions in molecular liquids and crystals, as well as homogeneous, surface, and enzymatic catalysis, scaling up to 100s-1000 of CC-level atoms[1,2,4-8]. We report unique datasets consisting of 10-100 gold standard interaction energies for drug-side chain[6], and protein pocket-cation[7]/drug[8] interactions between real-life drug molecules of 40-80 atoms and pocket models of 100-300 CC-level atoms.
[1] Chemical Science 15, 14556 (2024)
[2] MRCC program suite, www.mrcc.hu, JPCA 129, 2086 (2025)
[3] JPCA 128, 10282 (2024)
[4] JCTC 20, 7453 (2024)
[5] Nature Communication 12, 3927 (2021), J Am. Chem. Soc. 145, 25372 (2023)
[6] Puleva, Sanodas, Lőrincz, Charry, Rogers, Nagy, Tkatchenko (2025)
[7] Zhao, Lőrincz, Berta, Henkes, Nagy, Tkatchenko, Vuckovic (2025)
[8] Rezac, Lőrincz, Berta, Nagy (2025)
The Role of van der Waals Interactions on the Response Properties of Materials

Dr. Ariadni Boziki
Department of Physics and Materials Science, University of Luxembourg, Luxembourg City, Luxembourg.
Abstract
The accurate modeling of materials requires a careful treatment of van der Waals (vdW) interactions, which govern not only structural organization but also dynamic response properties. In this talk, I will examine how vibrational spectra and related observables in molecular crystals are shaped by dispersion forces, and how different theoretical models; including pairwise and many-body vdW corrections affect the predicted behavior. By systematically comparing results obtained with different dispersion-inclusive functionals, I will highlight the strengths and limitations of these methods in accurately capturing experimental observables. This discussion aims to underscore the necessity of an accurate treatment of vdW forces in modeling complex materials and guiding the design of new solids.
Structure and dynamics of weakly bound molecular clusters

Prof. Melanie Schnell
Deutsches Elektronen-Synchrotron DESY, Hamburg & Kiel University, Kiel, Germany.
Abstract
Rotational spectroscopy is a coherence technique known to determine the structures of molecules and molecular complexes in the gas phase in great detail – ranging from fundamental topics like proton transfer in microsolvated acids to astrochemistry. The structural information that can be deduced from the spectroscopic data provides an unprecedented molecular view on chemical processes, such as solvation and molecular recognition, governed by an interplay between hydrogen bonding and van-der-Waals interactions. For example, we studied complexes of polycylic aromatic hydrocarbons (PAHs), which are omnipresent in interstellar space, with an increasing number of water molecules to understand the initial steps of ice formation. We observe significant differences in the corresponding binding motifs for different PAHs, for example depending on the connectivity of the aromatic rings and the potential presence of hetero atoms. Furthermore, due to the high symmetry of the PAHs, rich intermolecular dynamics can be at play. In another example, we focused on water-driven dissociation of a single HCl molecule. In rotational spectroscopy, the nuclear spin of the chlorine atom is a sensitive probe of its electronic environment and thus its bonding situation. Consequently, our analysis allows us to precisely determine how many water molecules are needed for HCl to dissociate and provides insight into the intermolecular interactions of such complexes.
Van der Waals Force and Torque—Macroscopic and Microscopic Descriptions

Prof. Xiaofei Liu
State Key Laboratory of Mechanics and Control for Aerospace Structures, Key Laboratory for Intelligent Nano Materials and Devices of the Ministry of Education, Nanjing University of Aeronautics and Astronautics, Nanjing, China
Abstract
Van der Waals (vdW) interaction can be described either by microscopic theories such as ab initio quantum mechanical method and force-filed approximation or by macroscopic theories such as Lifshitz theory. It is highly desirable to achieve consistencies between the atomistic and continuum theories. Recent years, we revealed the dielectric function effect on the interlayer interaction of 2D materials and the vdW screening effect by graphene, by both theoretical and experimental investigations. Fifty years after the prediction of dielectric anisotropy-induced vdW torque by Parsegian et al., the effect can only be described by the continuum theory till now. We recently reproduced the vdW torque between dielectrically anisotropic 2D materials and related torque scaling laws using the fully atomistic many-body dispersion model. Also, we demonstrate a molecular simulation-compatible pairwise approximation that results in a dielectric function criterion of sign of vdW force formally identical to the Lifshitz criterion. Namely, the force computed from interatomic energy sum with materials’ and liquid’s C6 coefficients derived from Clausius-Mossotti relation will be repulsive, if liquid’s dielectric function ranks in-between those of unlike interacting materials.
Non-covalent interactions in molecular photocatalysis and photophysics

Prof. Christoph Bannwarth
Institute of Physical Chemistry, RWTH Aachen University, Aachen, Germany
Abstract
The ability to predict optical molecular properties and photochemical reaction outcomes using quantum chemistry methods becomes increasingly important for improving our mechanistic understanding and for advancing the computer-aided design of molecular emitters and photocatalysts. Key is the proper simulation of the photoinduced decay processes in the condensed phase.
In photocatalysis, the reaction pathways not only depend on the involved excited states, but also on the formation of a non-covalent complexes between the photocatalyst and the substrate. We will discuss a recently investigated photocatalytic cleavage reaction of chiral oxetanes. [1] Different cleavage pathways were found and shown to be dependent on the formation of stable non-covalent complexes between the catalyst and the substrate. Furthermore, the enantioselectivity can be rationalized from different association constants with the photocatalyst, the competition with cleavage products, and the substrate self-association.
Additionally, we will discuss the influence of excimer formation on the photoluminescence quantum yields (PLQYs) of luminescent organic molecules. Particularly, the formation of charge transfer states in these complexes can lead to significant reduction of the PLQY.[2]
We show that these processes can be modeled quite well using state-specifc orbital optimized density functional theory methods with dispersion corrections.
[1] N. Pflaum et al., J. Am. Chem. Soc., 2025, 147, 13893.
[2] M. Arnold et al., in preparation.
Neumann-Perrin dispersion correction. Twenty years of application in molecular crystal modelling

Dr. Dzmitry Firaha
Avant-garde Materials Simulation, Merzhausen, Germany
Abstract
Dzmitry Firaha, Subhayan Roychoudhury, Marcus Neumann
Accurate modelling of van der Waals interactions is essential for predicting the structure and stability of molecular crystals. The Neumann-Perrin (NP) dispersion correction [1], which is ne of the first physically grounded empirical based dispersion correction to the density functional theory, celebrates its 20 year anniversary this year. Originally parametrized in 2005, this correction has demonstrated robust performance for industrial crystal structure prediction (CSP) problems over the past two decades[2]. A key strength of the NP approach lies in its consistent reproduction of experimental crystal structures[3], which is an essential prerequisite for success in blind test CSP challenges[4]. The structures obtained with the NP correction have been used to calculate reliable and accurate free energies with TRHu(ST)23 method[5] to predict the stability of crystal forms under varying temperature and humidity conditions (Fig. 1). Our predictions align closely with experimental stability data, phase-transition temperatures and phase-transition relative humidities, highlighting the usefulness of these calculations to guide experimental screening and drug-formulation strategies.
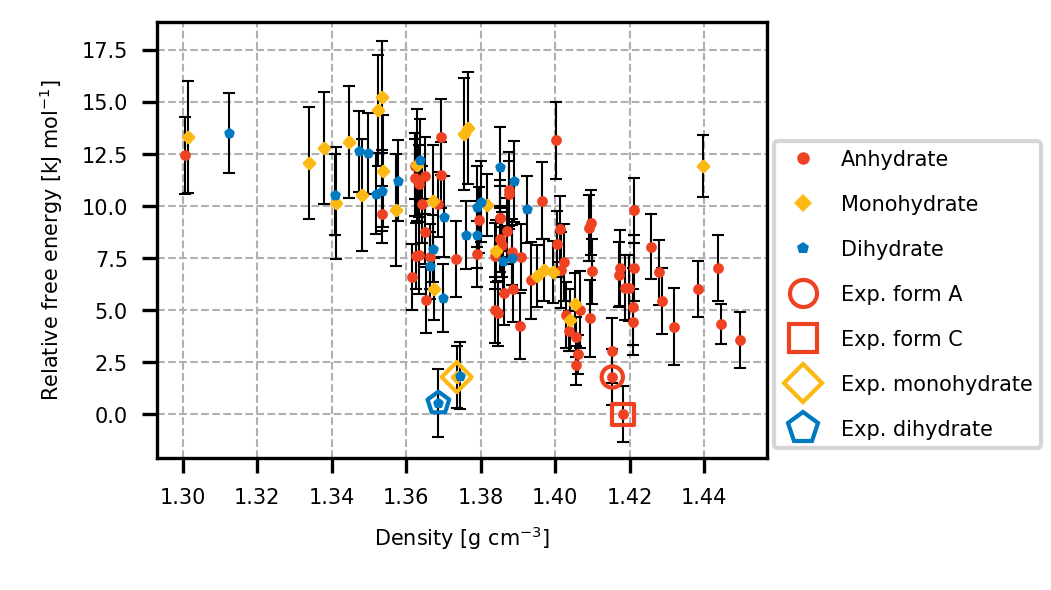
We demonstrate that the NP correction yields excellent agreement with experimentally determined lattice parameters and molecular arrangements across a broad range of organic crystals and compare it with other dispersion corrections models available to the community. These results underline the enduring value of the NP correction in bridging the gap between first-principles modelling and experimental crystallography, and in enabling in silico materials design starting from accurate dispersion modelling.
[1] Neumann, M. A., Perrin, M.-A. (2005), J. Phys. Chem. B, 109, 32, 15531.
[2] Neumann, M. A., et al. (2015), Nature Comm., 6.1, 7793; Hoja, J., et al. (2019), Sci. Adv. 5.1, eaau3338.
[3] van de Streek, J., Neumann, M. A. (2010), Struct. Sci., 66.5, 544; van de Streek, J., Neumann, M. A. (2014), Struct. Sci., 70.6, 1020.
[4] Day, G. M., et al. (2009). Struct. Sci., 65.2, 107; Bardwell, D. A., et al. (2011). Struct. Sci., 67.6, 535; Reilly, A. M., et al. (2016). Struct. Sci., 72.4, 439; Hunnisett, L. et al. (2024). Acta Cryst. B80, 517; Hunnisett, L. et al. (2024). Acta Cryst. B80, 548.
[5] Firaha, D. et al. (2023). Nature 623, 324.
Extending nonempirical DFT models to noncovalent weak interactions
Prof. Carlo Adamo
Chimie ParisTech, Institute of Chemistry for Life and Health Sciences, Paris, France
Abstract
Weak non-covalent interactions remain a challenging area for most Density Functional Approximations, and the inclusion of empirical potentials has proven to be an effective way to address their limitations. Recently, we have proposed an alternative approach based on the coupling of a non-empirical double hybrid functional, PBE-QIDH, with a specifically developed basis set named DH-SVPD. This basis set was constructed using a dedicated procedure that does not involve any fitting to external reference data [1]. The results obtained across a wide range of benchmark datasets [2-4] demonstrate that our protocol, PBE-QIDH/DH-SVPD, provides interaction energies that are at least as accurate as those obtained with comparable functionals combined with large triple- or quadruple-ζ basis sets and dispersion corrections. The rationale behind this performance can be attributed to improvements in the description of electronic molecular properties induced by our protocol.
Acknowledgement. Funded by the European Union (ERC, project MaMa, n. 101097351). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.
[1] P J. S. Garcı́a, É. Brémond, M. Campetella, I. Ciofini, C. Adamo, J. Chem. Theory Comput., 2019, 15, 2944–2953.
[2] E. Brémond, H. Li, J. C. Sancho-García, C. Adamo J. Phys. Chem. A, 2022, 126, 2590–2599.
[3] H. Li, L. Briccolani-Bandini, B. Tirri, G. Cardini, E. Brémond, J. C. Sancho-García, C. Adamo, J. Phys. Chem. A, 2024, 128, 6581–6592.
[4] H. Li, E. Brémond, J. C. Sancho-García, Á. J. Pérez-Jiménez, G. Scalmani, M. J. Frisch and C. Adamo, Phys. Chem. Chem. Phys., 2024, 26, 8094–8105.
Assessing van der Waals interactions in hybrid systems made of biomolecules and materials
Prof. Maria Fyta
Computational Biotechnology, RWTH Aachen University, Aachen, Germany
Abstract
Taner Dogan, Lydia Spatz, Peijia Wei, Longlong Li, Prof. Maria Fyta
VHybrid systems made of a biomolecular and a material part, typically, within an electrolyte solution are involved in complex interactions. An important component in these interactions are dispersion or van der Waals forces and are not always easy to handle and model. The strength of these interactions is strongly dependent on the molecular and material type and their relative arrangement. This rich dependence is not always sufficiently taken into account by interaction models. In order to understand these differences and assess the quality of respective models, we turn to density functional theory (DFT) and choose different schemes for van der Waals corrected exchange correlation (vdw-XC) functionals. Where necessary we complement the insights gained with those from atomistic simulations. As probes for the assessment of the vdw-XC functionals, we model two types of cases reflecting different interaction and bonding environments: (a) single amino acids placed within a graphene pore and (b) single amino acids and sugar molecules interacting with defective graphene. The interaction curves extracted from the respective DFT simulations are assessed providing important ideas towards the modeling of van der Waals interactions in hybrid bio/material systems with applications in protein detection and biosensing.
On the mapping of multi-dimensional potential energy surfaces of weakly bound complexes of increasing size: state-to-the-art and challenges
Prof. Majdi Hochlaf
Université Gustave Eiffel, COSYS/IMSE, Champs sur Marne, France.
Abstract
To treat the spectroscopy and dynamics of small and medium-sized weakly bound complexes, one needs an accurate multi-dimensional potential energy surface (mD-PESs). To date, first principles methodologies are available for the generation of such precise mD-PESs. They include Coupled Clusters methods extrapolated to complete basis set limit and where diverse non adiabatic and relativistic corrections are considered. Nevertheless, these methods can be used mostly for small molecular systems (i.e. di, tri or tetratomics) since their computational costs become prohibitive while increasing the size of the molecular systems. One needs alternative approaches where the computational cost is reduced without deteriorating the accuracy. For those purposes, several approaches were developed making use of explicitly correlated methodologies, or of composite schemes where we combine DFT/MP2 methods with either standard or explicitly correlated techniques, or of SAPT based methods, and of machine learning techniques.
Several examples will be used presented and discussed.
Collective Interactions
Prof. Cina Foroutan-Nejad
Institute of Organic Chemistry, Polish Academy of Science, Warsaw, Poland.
Abstract
A century after G. N. Lewis introduced his foundational model of chemical bonding, its core principles remain central to our understanding of molecular structure. Traditionally, neighboring atoms in a Lewis structure are assumed to share stabilizing interactions. However, this assumption begins to falter at the boundary between covalent and noncovalent bonding. Our work reveals that, in certain molecules with the general formula MAX₃—where M is a metal, A a nonmetal, and X an electron-rich or electron-withdrawing group—the interaction between adjacent M and A atoms can be destabilizing. Remarkably, the overall structure is preserved due to strong interactions between electron-deficient metals and the X groups.1–6 This phenomenon gives rise to unconventional structures, such as the inverted LiCF₃, where lithium interacts preferentially with fluorine atoms over the central carbon in the gas phase. These collective interactions extend beyond organometallic compounds and offer insight into anomalous bond dissociation energies and stability trends across a wide range of molecules—from perhaloalkanes to the elusive cyclic C₆O₆. Our computational studies highlight the broad relevance of this concept in modern chemical bonding theory.
1. Foroutan‐Nejad, C. The Na⋅⋅⋅B Bond in NaBH3−: A Different Type of Bond. Angew. Chem. Int. Ed. 59, 20900–20903 (2020).
2. Sowlati-Hashjin, S. et al. Collective interactions among organometallics are exotic bonds hidden on lab shelves. Nat. Commun. 13, 2069 (2022).
3. Šadek, V. et al. Reply to: On the existence of collective interactions reinforcing the metal-ligand bond in organometallic compounds. Nat. Commun. 14, 3873 (2023).
4. Badri, Z. & Foroutan-Nejad, C. Classical versus Collective Interactions in Asymmetric Trigonal Bipyramidal Alkaline Metal‒Boron Halide Complexes. Chem. – Eur. J. n/a, e202400156 (2024).
5. Pino-Rios, R., Báez-Grez, R. & Foroutan-Nejad, C. Anti-electrostatic cation⋯π-hole and cation⋯lp-hole interactions are stabilized via collective interactions. Chem. Commun. 60, 400–403 (2024).
6. Bandeira, N. A. G., Martín Pendás, Á. & Foroutan-Nejad, C. Reply to: An approach to the resolution of the dispute on collective atomic interactions. Nat. Commun. 15, 10403 (2024).
Grid-projected force-fields for high throughput simulations of molecules on surface
Dr. Prokop Hapala
FZU - Institute of Physics of the Czech Academy of Sciences, Prague, Czech Republic.
Abstract
Prokop Hapala (FZU - Institute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, Prague 8, 182 00, Czech Republic),
Paolo Nicolini (FZU - Institute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, Prague 8, 182 00, Czech Republic),
Indranil Mal (FZU - Institute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, Prague 8, 182 00, Czech Republic),
Milan Kocí (FZU - Institute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, Prague 8, 182 00, Czech Republic),
Niko Oinonen (Aalto University, Department of Applied Physics, P.O. Box 11000, Otakaari 1B, AALTO, FI-00076, Finland),
Aliaksandr V. Yakutovich (nanotech@surfaces laboratory, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, Dübendorf, CH-8600, Switzerland),
Aurelio Gallardo (IMDEA Nanoscience Institute, C/ Faraday 9, Campus de Cantoblanco, Madrid, 28049, Spain),
Martin Ondráček (FZU - Institute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, Prague 8, 182 00, Czech Republic),
Ondřej Krejčí (Aalto University, Department of Applied Physics, P.O. Box 11000 (Otakaari 1B), AALTO, FI-00076, Finland)
Rapid exploration of molecular configurations on surfaces is crucial for the design and rationalization of self-assembled structures, as well as for simulations of molecular manipulation using scanning probe microscopy - both essential for advancing molecular electronics and nanotechnology. The computational bottleneck in these simulations typically lies in evaluating non-covalent interactions between molecules and substrates, even when using simple pairwise potentials such as Lennard-Jones or Morse.
To address this challenge, we developed the Grid-Projected Force Field (GridFF) method, which leverages the rigidity of the substrate by replacing explicit pairwise non-covalent interactions between atoms with interpolation from a precomputed grid. This not only accelerates simulations by 1–2 orders of magnitude but also enables the use of more sophisticated interaction potentials at virtually no additional computational cost.
We have implemented GridFF in two high-throughput simulation packages. The first, ppafm [1], simulates 3D stacks of high-resolution atomic force microscopy (AFM) images (~1 million pixels) in less than 0.1 seconds, with accuracy comparable to density functional theory (DFT). This is achieved using GridFF constructed from convolution of ab initio electron densities and Grimme D3 van der Waals corrections. The ppafm package has become a primary tool for generating training data for machine-learning-based reconstruction of molecular structures from AFM images [2].
The second application is FireCore [3], a newly developed package for configuration sampling of small organic molecules on surfaces. FireCore exploits GPU parallelism to run thousands of system replicas simultaneously, enabling the sampling of ~15 million configurations per second of a xylitol molecule on a NaCl substrate using a single desktop GPU. We envision FireCore playing a role in training autonomous nanomanipulation protocols analogous to the role ppafm plays in imaging. To further enhance the accuracy of these simulations, we are currently integrating efficient hydrogen-bond corrections into the GridFF framework.
[1] Oinonen, N. et al. Advancing scanning probe microscopy simulations: A decade of development in probe-particle models. Computer Physics Communications 305, 109341 (2024).
[2] Alldritt, B. et al. Automated structure discovery in atomic force microscopy. Sci. Adv. 6, (2020).
[3] https://github.com/ProkopHapala/FireCore
Anharmonic Intermolecular Vibrations of Molecular Crystals via Vibrational Perturbation Theory
Dr. Johannes Hoja
Department of Chemistry, University of Graz, Graz, Austria.
Abstract
Low-frequency vibrations of molecular crystals are extremely challenging to describe, but are the most crucial part for determining vibrational entropy and hence free energies at finite temperatures, often leading to qualitative changes in the stability ordering of polymorphs [1]. Furthermore, THz spectroscopy enables the distinction between such different crystal-packing arrangements of the same molecule. These low-frequency vibrations are collective lattice modes representing intermolecular translations and rotations and are often dominated by van der Waals dispersion interactions. Therefore, an accurate account of dispersion interactions in the utilized electronic structure method is essential. In terms of the vibrational description, the harmonic or quasi-harmonic approximation is often insufficient given the anharmonic nature of many intermolecular modes. We have recently shown that a very good description of experimental fundamental frequencies, within about 6 cm-1 on average, can be obtained for molecular dimers by using second-order vibrational perturbation theory (VPT2) combined with hindered-rotor models together with CCSD(T) calculations [2].
Herein, we introduce a VPT2 approach that can be applied to periodic molecular crystal structures. Anharmonic force constants are calculated for monomers and relevant dimers of the respective crystal, which are then embedded into fully periodic harmonic calculations using a multimer embedding scheme [3]. We illustrate this approach for small dispersion-bound molecular crystals evaluated at the PBE+MBD level with a special focus on the accuracy of low-frequency intermolecular vibrations. This approach also yields improved free energies that are vital for calculating lattice energies or energy barriers for transformations between molecular crystal polymorphs [4], for which dispersion interactions play an imperative role as well. Compared with canonical fully periodic VPT2 calculations, the introduced fragment approach is far more cost-effective and paves the way toward highly accurate predictions of THz spectra and vibrational free energies for molecular crystals.
[1] J. Hoja, H.-Y. Ko, M. A. Neumann, R. Car, R. A. DiStasio Jr., A. Tkatchenko, Sci. Adv. 5, aau3338 (2019).
[2] J. Hoja, A. D. Boese, J. Chem. Phys. 161, 234110 (2024).
[3] J. Hoja, A. List, A. D. Boese, J. Chem. Theory Comput. 20, 357–367 (2024).
[4] N. Goncharova, J. Hoja, arXiv preprint, arXiv:2410.10506 (2025).
Protein–membrane interactions with a twist
Dr. Martin Michael Müller
Université de Lorraine, CNRS, LPCT, France.
Abstract
Jordan Klein, Loréne Schad, Thérèse E. Malliavin, Martin Michael Müller
To understand how a biofilament can interact with a lipid membrane, mesoscopic models are of particular interest. Within a framework of elasticity theory and geometry, two mechanisms have been proposed, which can induce membrane deformations due to a biofilament: the Twister and the Darboux torque mechanism [1]. Whereas the Darboux torque mechanism has been shown to explain membrane deformations by a polymer in several important biological systems, the Twister mechanism has been studied more carefully only recently, which has allowed for the development of a possible explanation for the membrane translocation of botulinium toxins [2].
In my talk I will present coarse-grained molecular dynamics simulations of a protein in interaction with a membrane. The protein is modeled as a cylinder stabilized by a tensegrity scheme, leading to an elasticity similar to that observed in real proteins. The Twister mechanism is induced by a hydrophobic helical strip displayed by the protein. The entire configuration space is explored by systematically varying the hydrophobic strip width, the twisting of the strip as well as the range of hydrophobic interactions between the protein and the membrane. The results are explained using a qualitative model based on the total hydrophobic moment of the protein.
[1] J. Fierling, Albert Johner, Igor M. Kulić, Hervé Mohrbach, Martin Michael Müller. Soft Matter, 12:5747, 2016.
[2] A. Delort, Grazia Cottone, Thérèse E. Malliavin, Martin Michael Müller. IJMS, 25:2481, 2024.
[3] Jordan Klein, Loréne Schad, Thérèse E. Malliavin, Martin Michael Müller. Soft Matter, 21: 4336, 2025.
Chemical reactivity from a combined Conceptual DFT and ELF topology approach
Bastien Courbiere
Laboratoire de Chimie Théorique, Sorbonne Université, Paris, France.
Abstract
B. Courbière, J. Pilmé
Laboratoire de Chimie Théorique, Sorbonne Université, Paris 75005, France.
Unravelling chemical reactivity can be compared to solving a puzzle: understanding how molecules interact, reorganize, and evolve during a reaction requires both empirical insight and intuition. The study of reactivity, developed to assist experimentalists and provide rationalization tools, is a well-established field in molecular modelling. However, most approaches rely on computationally demanding methods or require long calculation times. This project introduces an innovative method to predict the energetic profile of chemical reactivity, from separated reactants to transition states, at a low computational cost. By combining the Dual Descriptor (DD)[1], from Conceptual DFT[2-3], condensed within the Electron Localization Function (ELF)[4] topology, we propose an approach that captures early-stage interactions while mimicking the DFT energies. Our approach builds on the Klopman-Salem[5-6] model, which isolates Coulomb contributions between molecular fragments treated as monomers.
The so-called "EDD", depends entirely on the relative orientation between the “frontier” densities of molecular fragments. By integrating ELF probability indicators within Dual Descriptor domains, this framework[8] offers a fast, cost-effective and computationally accessible alternative to traditional methods, providing a reliable and efficient way to understand and predict chemical reactivity. We illustrate this approach on several prototypical reactions, from simple non-covalent interactions to intricate reactivity profiles.
[1] C. Morell, A. Grand, A. Toro-Labbé, New Dual Descriptor for Chemical Reactivity, J. Phys. Chem. A 109, 205–212 (2005)
[2] W. Yang, R. G. Parr, Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis, Proc. Natl. Acad. Sci. U. S. A. 82, 6723–6726 (1985)
[3] R. G. Parr, W. Yang, Density-Functional Theory of the Electronic Structure of Molecules, Annu. Rev. Phys. Chem. 46, 701–728 (1995)
[4] B. Silvi, A. Savin, Classification of Chemical Bonds Based on Topological Analysis of Electron Localization Functions, Nature 371, 683–686 (1994)
[5] L. Salem, Intermolecular Orbital Theory of the Interaction between Conjugated Systems. I. General Theory, J. Am. Chem. Soc. 90, 543–552 (1968)
[6] G. Klopman, Chemical Reactivity and the Concept of Charge- and Frontier-Controlled Reactions, J. Am. Chem. Soc. 90, 223–234 (1968)
[7] J. Klein, P. Fleurat-Lessard, J. Pilmé, New Insights in Chemical Reactivity from Quantum Chemical Topology, J. Comput. Chem. 42, 840–854 (2021)
[8] B. Courbière, J. Pilmé, Exploring Chemical Reactivity through a Combined Conceptual DFT and ELF Topology Approach, J. Mol. Model. 30, 362 (2024)
ADLD and ADEX: New Lenses for Atomic-Level Analysis of Non-Covalent Interactions
Gianluca Regni
University of Perugia, Perugia, Italy.
Abstract
We present a novel quantum chemical method for quantifying atomic contributions to London dispersion (LD) energy in molecular systems, with a “gold standard” accuracy within the Local Energy Decomposition framework. This method, called Atomic Decomposition of London Dispersion energy (ADLD), decomposes LD interactions into atom-wise contributions, providing a clearer and more interpretable picture of how dispersion affects each part of the molecule. This level of resolution is especially powerful for large and complex systems, where it is often difficult to intuitively identify which functional groups contribute most to the total dispersion energy. ADLD also makes it possible to observe how London dispersion responds to variations in electronic structure, such as spin state, charge, and resonance, directly at the atomic level, offering valuable insights for molecular design in both drug discovery and materials science.
In addition to ADLD, we also present the Atomic Decomposition of EXchange energy (ADEX) method, which provides an analogous atom-wise partitioning for the exchange energy within the LED framework. Together, ADLD and ADEX help provide atom-wise insights into key components of non-covalent interactions within molecular systems, enhancing our ability to analyze and design complex molecules.